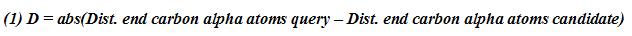| METHOD |
- Search Space
The Search Space is a multidimensional library of loops of know structures
organized into three a three levels hierarchy:
(i) at the top, loops are
identified according to the type of the bracing secondary structures:
αα loops, βα loops, αβ loops and ββ loops;
(ii) at the next level, loops are grouped
according to their length, and finally
(iii) loops are grouped according to
the geometry of bracing secondary structures.
This geometry of a loop is defined by: a distance, D, and three angles:a hoist (δ),
a packing (θ) and a meridian (ρ)[3]
(Figure 1).
|
ALGORITHM
The prediction algorithm selects a set of candidate loops from the Search Space,
then subsequently filters and ranks them by various criteria
(Figure 1).
- Selection
The Search Space is queried by the length of the loop,
the type of secondary structures that span the query loop and
by the geometry of the motif. Loops with the same length (+/- 1 residue)
that belong to the same geometrical bin are selected.
Two loops (loop A and B with geometry GA=(DA,δA,θA,σA)
and GB=(DB,δB,θB,σB), respectively), share the same geometry if the difference in
geometry falls inside the semi-open interval [(0,0,0,0),(2,30,30,45)).
- Filtering
In the filtering step in the algorithm discards clearly unfavorable
candidates by assessing the fit of stem regions and by steric fitting in
the new protein framework. The terms of steric violations or clashes are
computed among main chain atoms (N, C, Cα and O). Two atoms are in steric
clash if their distance is smaller than the 70% of sum of the respective
van der Waals radii.
- Ranking
The final set of candidate loops are ranked by two measures:
(1) A sequence similarity score between the query and candidate loops
using the conformation similarity weight matrix (K3 matrix)[3];
(2) Φ/φ main chain dihedral angle propensities. The dihedral angle
propensity score measures the compatibility of observed and expected dihedral
angles of each residue of the candidate loop in the corresponding position
of the query. Main chain conformation definitions and propensities are
defined according to the p15 propensities table of D. Shortle[4]
The two components of the scoring scheme, sequence and propensity,
are combined into a composite Zscore.
|
REFERENCES
- 1. A. A. Canutescu, A. A. Shelenkov, and R. L. Dunbrack, Jr. A graph
theory algorithm for protein side-chain prediction.
Protein Science 12, 2001-2014 (2003).
- 2. A. Sali & T.L. Blundell. Comparative protein modelling by satisfaction
of spatial restraints. J. Mol. Biol. 234, 779-815, (1993).
- 3. B. Oliva, PA. Bates, E. Querol, FX. Aviles, MJ. Sternberg. An automated
classification of the structure of protein loops. J. Mol. Biol. 266, 814-830 (1997).
- 4. A.S. Kolaskar and U. Kulkarni-Kale. Sequence alignment approach to
pick up conformationally similar protein fragments.
Journal of Molecular Biology 223, 1053-1061 (1992).
- 5. D. Shortle. Composite of local structure propensities:
evidence for local encoding of long-range structure.
Protein Science 11, 18-26 (2002).
|